MSigDB SQLite Database
Introduction#
With the release of MSigDB 2023.1 we have created a new SQLite database for the fully annotated gene sets in both the Human (2023.1.Hs) and the Mouse (2023.1.Ms) resources. Each ships as a single-file database usable with any compliant SQLite client. No other downloads are necessary. This new format provides the MSigDB contents and metadata with all of the searchability and manipulative power of a full relational database. Like the XML format that has been made available since the early days of MSigDB, the SQLite format has the advantage of being self-contained and portable and thus easy to distribute, archive, etc. In addition, the SQLite format allows us to open up the data to ad-hoc SQL queries.
Note that we will continue producing the XML file for now, but it should be considered deprecated with the intention to eventually be entirely removed in a future release.
Below we describe the design of the MSigDB relational database and provide some examples of useful SQL queries. General information about SQLite can be found at the end of this document.
The License Terms for MSigDB are available on our website.
Database Design#
Design Considerations#
The schema is designed to be easy and (reasonably) fast for end-users. We decided that some amount of denormalization (e.g. the collection_name and license_code columns on the gene_set table) makes the database easier to understand and use.
Similarly, we wanted to prevent extraneous information from causing the design to be more difficult to use. Thus, each database file will hold only ONE MSigDB release for ONE resource, either Human or Mouse, with very little in the way of history tracking. It was necessary to ship the resources separately to prevent conflicts between them (there are gene sets in both with identical names, for example), but doing so also simplifies their use.
This schema is designed to be a read-only resource. After an MSigDB version is released it doesn't change. Any changes mean a new version. Notably, this allows us to side-step the known limitations and potential issues of using SQLite in the context of multiple concurrent writers. These simply do not apply other than during initial creation. SQLite has no issues around multiple concurrent readers.
Schema#
Referring to the schema diagram below, the tables in blue are core to defining the gene sets and the genes they contain, while those in purple provide the metadata about the gene sets, the genes, and MSigDB itself. The tables in gray give data about gene sets that were considered for, but excluded from, the MSigDB release, as explained below.
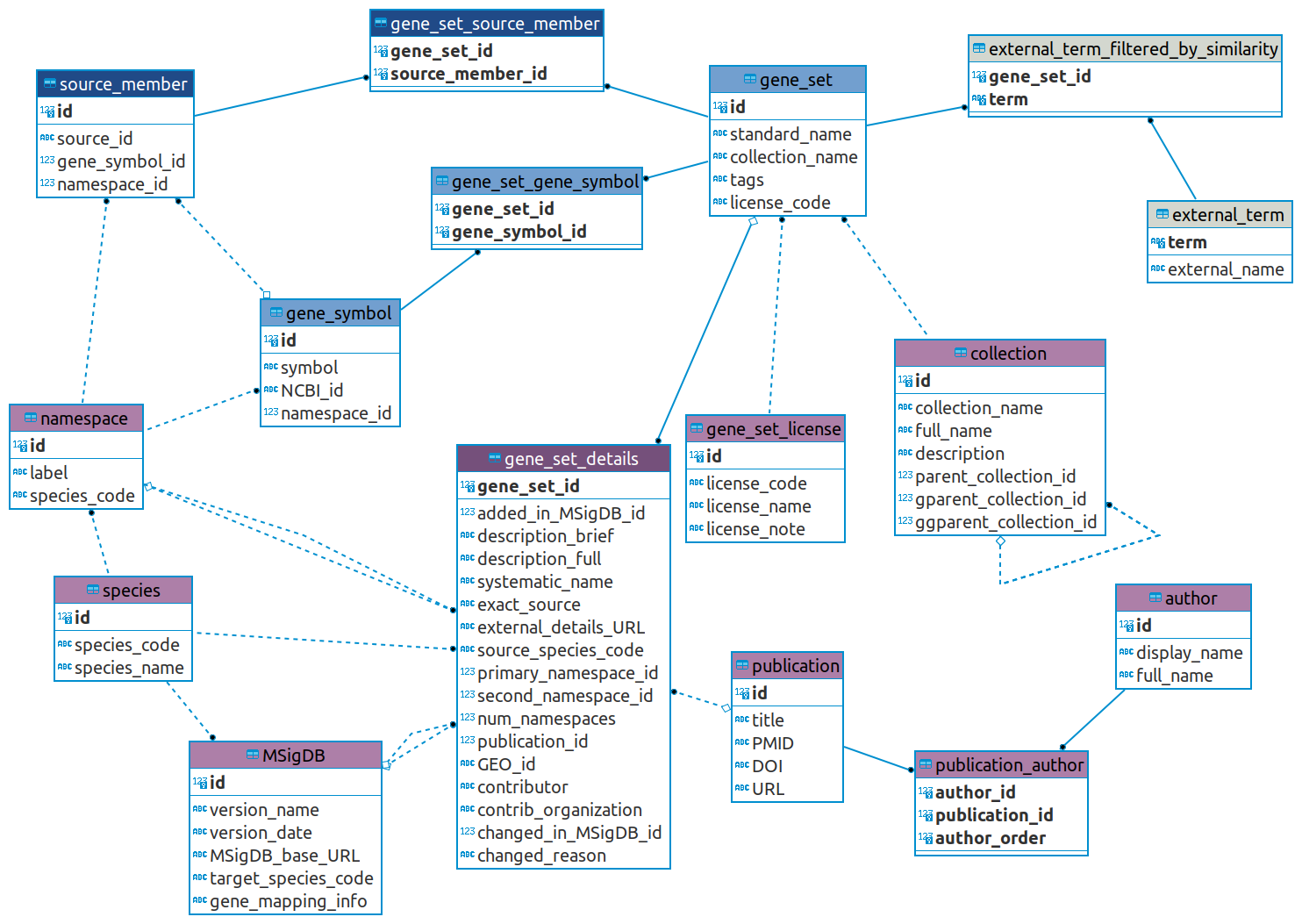
Note that in all cases of tables with an id primary key column, these primary key values are generated synthetically and will not be considered stable across different versions of MSigDB (and likewise when used as a foreign key). In other words, the id of a particular gene set, gene symbol, author, etc. will likely have a different value in the next version of MSigDB. While usable within a given database for JOIN queries and so on, these values should not be relied upon outside of that context.
The core (blue) tables:
- The gene_set table holds the core information about each gene set. Note that the collection_name and license_code columns are denormalized for ease of use; these hold the name of the MSigDB collection and its license respectively.
- The tags column is unused at present and reserved for future use. It may be removed in the future in favor of a more structured alternative for providing tag metadata.
- The gene_symbol table holds the canonical information for the genes found in MSigDB gene sets, including both the official symbol (HUGO for Human MSigDB, MGI for Mouse) and the NCBI (formerly Entrez) Gene ID. The namespace_id will be constant across a given database as all symbols are mapped into the same namespace for a particular release of MSigDB.
- The gene_set_gene_symbol table joins the gene sets to its member gene symbols.
- In addition to the canonical gene symbols, which are in the same namespace across all gene sets in an MSigDB release, all gene sets include the gene identifiers of its members as specified by the original source of the gene set. This original source will commonly be a publication, for example, or some broader resource like Reactome or Gene Ontology. The source_member table contains these original gene set member identifiers (joined via gene_set_source_member).
- The gene_symbol_id column gives the mapping to our uniformly mapped gene symbols. We provide a set of external CHIP files encoding the same information which will usually be more convenient to use, however.
- These tables should not be used when using the database to extract gene sets for custom gene set files for use with GSEA and other analysis tools as the source identifiers will not have a uniform namespace, may conflict with one another, and may not even have a valid mapping in modern namespaces. These tables are meant for informational purposes only.
The metadata (purple) tables:
- The gene_set_details table gives a variety of additional details for each gene set. It is essentially an extension of the core gene_set table - and uses the same primary key - but is kept separate in order to simplify the core table.
Here are some columns of note:- While each database of MSigDB is targeted at a particular species (Human or Mouse), the members of a given gene set may have originated in a different species than the target. This is given in the source_species_code column.
- The external_details_URL column may actually contain multiple URLs. These will be separated by the pipe character ('|').
- The exact_source column holds information on finding the source of the gene set from wherever it originated. For external resources like Reactome or Gene Ontology this is frequently an identifier defined by the resource itself (e.g. R-HSA-156588) which can be used to look up further details on that resource's website. The column can also hold free-text listing e.g. a figure, section or supplementary document from a publication.
- While we now require all new gene sets to consist of members from a single namespace, some older sets contain members from a mix of namespaces. These are found in the primary_namespace_id, secondary_namespace_id, and their count in num_namespaces. For the relatively few cases where there are more than two, any additional namespaces can be found by iterating through the linked source members.
- The added_\in_MSigDB_id, changed_in_MSigDB_id, and changed_reason columns are unused at present and reserved for future use. They are intended to hold MSigDB revision history.
- The collection table holds the information for each MSigDB Collection. For convenience, the collection_name column encodes the full collection hierarchy information, in the form "C5:GO:BP" or "M2:CP:REACTOME" for example. There is also a fully recursive hierarchy encoded in the table but we expect few users to need this.
- The gene_set_license table allows us to associate licensing info with each gene set. The vast majority are Creative Commons Attribution 4.0 International (CC-BY-4.0); see our License Terms page for more info.
- The MSigDB table gives information about the database as a whole. It contains information about the date of release, the mapping information used (where available), the target species, etc. There are records covering all versions of MSigDB going back from the current version to the original 1.0 release. While these older records are not currently referenced, they are included to cover the future intent to add revision history in the added_in_MSigDB_id and changed_in_MSigDB_id columns of the gene_set_details table as mentioned earlier.
- The namespace and species tables allow us to label source_member and gene_symbol records to identify the mapping info associated with each (that is, what kind of identifier or symbol we have), as well as the overall target species of MSigDB itself. Note again that the source identifier of a particular gene set member might differ from the MSigDB target species.
- The publication and author tables associate publication info to gene sets (joined by publication_author). Where possible, we have extracted the author name info from PubMed based on the PubMed ID (PMID). This is imperfect, however, as there are cases of distinct authors with identical names. Our information here is only as good as PubMed allows it to be. Be sure to reference the publication itself for the most accurate authorship info. There are a few cases of gene sets with author info but without an associated publication in PubMed. These are represented through "placeholder" publication records with titles like "Placeholder publication for M2872,M2873", where the identifiers at the end are the systematic_name(s) of the corresponding gene set.
The "external item" (gray) tables:
- When mining external resources for gene sets, e.g., Reactome, Gene Ontology, Human Phenotype Ontology, we sometimes find that the resulting collection would contain multiple gene sets that are too similar if we include them all. We apply a redundancy filtering procedure and select a single representative of similar candidate gene sets and exclude the others. MSigDB’s online gene set page of a selected gene set includes information about any related candidate gene sets that were excluded, linking out to details on the external resource’s website. The gray tables external_term and external_term_filtered_by_similarity contain this information.
Example Queries#
The examples given here assume we are working with the MSigDB Human database from our Downloads page (msigdb_v2023.1.Hs.db is the current version at the time of this writing). Note that we ZIP the database to reduce its size, so you must decompress it first before use.
These examples also assume the use of the official SQLite command line shell to keep everything consistent across all platforms. The exact results may vary depending on the version of the database you are using and the particular query.
NOTE: there is a known issue where extraneous records were retained in certain tables in the MSigDB 2023.1.Hs and 2023.1.Mm release. While that won't affect the flow of the following discussion, we recommend addressing it before using the databases beyond the given examples. Please refer to those Release Notes for specific instructions.
Extracting gene sets in the GMT format#
One key use-case for performing SQL queries against the database involves building custom collections of gene sets, so those have been designed to be fast and convenient. For example, the following will select all the WikiPathways sets in the Human database into a GMT file named wikipathways.gmt:
.mode tabs
.once wikipathways.gmt
SELECT standard_name 'na', group_concat(symbol, ' ')
FROM gene_set gset
INNER JOIN gene_set_gene_symbol gsgs on gset.id = gene_set_id
INNER JOIN gene_symbol gsym on gsym.id = gene_symbol_id
WHERE collection_name = 'C2:CP:WIKIPATHWAYS'
GROUP BY standard_name ORDER BY standard_name ASC;
The basic template for creating GMTs is as follows:
.mode tabs
.once <filename>
SELECT standard_name 'na', group_concat(symbol, ' ')
FROM gene_set gset
INNER JOIN gene_set_gene_symbol gsgs on gset.id = gene_set_id
INNER JOIN gene_symbol gsym on gsym.id = gene_symbol_id
WHERE <selection criteria>
GROUP BY standard_name ORDER BY standard_name ASC;
Simply vary the criteria in the WHERE clause to determine the contents of the output GMT. The first two lines are SQLite specific directives (fill in the desired file name on line 2). Note that the second argument to the group_concat function is a quoted tab character.
Finding gene sets containing one or more specified genes#
Here's another simple example that finds the names of all gene sets which have BRCA1 or BRCA2 as a member:
SELECT distinct(standard_name)
FROM gene_set gset
INNER JOIN gene_set_gene_symbol gsgs ON gset.id = gsgs.gene_set_id
INNER JOIN gene_symbol gsym ON gsym.id = gsgs.gene_symbol_id
WHERE symbol in ('BRCA1', 'BRCA2') ORDER BY standard_name;
AAAYWAACM_HFH4_01
ACTAYRNNNCCCR_UNKNOWN
ACTGAAA_MIR30A3P_MIR30E3P
ARID3B_TARGET_GENES
ASH1L_TARGET_GENES
<...etc...>
Extracting gene sets and their metadata#
This query gets all the Reactome sets after applying a size threshold of between 15 and 500 genes. Here we are also providing a full link to the gene set on the GSEA-MSigDB website in place of the ‘na’ of the earlier example:
.mode tabs
.once wikipathways_threshold.gmt
SELECT standard_name,
( SELECT MSigDB_base_URL FROM MSigDB WHERE version_name = '2023.1.Hs' )
||'/geneset/'||standard_name,
group_concat(symbol, ' ')
FROM gene_set gset
INNER JOIN gene_set_gene_symbol gsgs on gset.id = gene_set_id
INNER JOIN gene_symbol gsym on gsym.id = gene_symbol_id
WHERE collection_name = 'C2:CP:WIKIPATHWAYS'
GROUP BY standard_name HAVING count(symbol) BETWEEN 15 AND 500
ORDER BY standard_name ASC;
Note that here we are using a subquery to get the MSigDB_base_URL to build the website link:
SELECT MSigDB_base_URL FROM MSigDB WHERE version_name = '2023.1.Hs'
This next query builds on our earlier example combined with the above to get all sets with either BRCA1 or BRCA2 as a member in that size range and save them to a GMT:
.mode tabs
.once BRCA1_BRCA2_sets.gmt
SELECT standard_name,
(SELECT MSigDB_base_URL FROM MSigDB WHERE version_name = '2023.1.Hs')
||'/geneset/'||standard_name,
group_concat(symbol, ' ')
FROM gene_set gset
INNER JOIN gene_set_gene_symbol gsgs on gset.id = gene_set_id
INNER JOIN gene_symbol gsym on gsym.id = gene_symbol_id
WHERE gset.id IN
( SELECT distinct(gene_set_id)
FROM gene_set_gene_symbol gsgs2
INNER JOIN gene_symbol gsym2 ON gsym2.id = gsgs2.gene_symbol_id
WHERE symbol in ('BRCA1', 'BRCA2') )
GROUP BY standard_name HAVING count(symbol) BETWEEN 15 AND 500
ORDER BY standard_name ASC;
This query gets some more detailed information about a particular named gene set, including the PubMed ID:
.mode tabs
.headers on
SELECT collection_name, license_code, PMID AS PubMedID, description_brief
FROM gene_set gset
INNER JOIN gene_set_details gsd ON gsd.gene_set_id = gset.id
INNER JOIN publication pub ON pub.id = publication_id
WHERE standard_name = 'ZHOU_CELL_CYCLE_GENES_IN_IR_RESPONSE_6HR';
collection_name license_code PubMedID description_brief
C2:CGP CC-BY-4.0 17404513 Cell cycle genes significantly (p =< 0.05) changed in fibroblast cells at 6 h after exposure to ionizing radiation.
Now, get the Title and Authors for the PubMed ID from the above:
SELECT title, group_concat(display_name) AS Authors
FROM publication pub
INNER JOIN publication_author pa ON publication_id = pub.id
INNER JOIN author au ON author_id = au.id
WHERE PMID = 17404513;
title Authors
Identification of primary transcriptional regulation of cell cycle-regulated genes upon DNA damage. Zhou T,Chou J,Mullen TE,Elkon R,Zhou Y,Simpson DA,Bushel PR,Paules RS,Lobenhofer EK,Hurban P,Kaufmann WK
This query will find the External Term(s) and Name(s) that were filtered out as similar by our redundancy check for a given GOBP gene set:
SELECT et.term, external_name
FROM external_term et
INNER JOIN external_term_filtered_by_similarity etfbs ON etfbs.term = et.term
INNER JOIN gene_set gset ON gset.id = etfbs.gene_set_id
WHERE standard_name = 'GOBP_MITOTIC_SPINDLE_ELONGATION';
term external_name
GO:0051256 mitotic spindle midzone assembly
Extracting a summary of gene sets#
This query will extract a summary of selected gene sets with a short example WHERE clause to restrict it to the C5:GO collection only. You can add a more detailed WHERE clause and the column selection can be expanded or reduced as desired:
SELECT standard_name, count(gene_symbol_id), collection_name,
source_species_code, ns.label, contributor, PMID
FROM gene_set gset
INNER JOIN gene_set_details gsd ON gsd.gene_set_id = gset.id
INNER JOIN namespace ns ON ns.id = primary_namespace_id
LEFT JOIN publication pub ON publication_id = pub.id
INNER JOIN gene_set_gene_symbol gsgs ON gsgs.gene_set_id = gset.id
WHERE collection_name LIKE "C5:GO:%"
GROUP BY standard_name ORDER BY standard_name limit 3;
standard_name count(gene_symbol_id) collection_name source_species_code label contributor PMID
GOBP_10_FORMYLTETRAHYDROFOLATE_METABOLIC_PROCESS 6 C5:GO:BP HS Human_NCBI_Gene_ID Gene Ontology
GOBP_2FE_2S_CLUSTER_ASSEMBLY 11 C5:GO:BP HS Human_NCBI_Gene_ID Gene Ontology
GOBP_2_OXOGLUTARATE_METABOLIC_PROCESS 17 C5:GO:BP HS Human_NCBI_Gene_ID Gene Ontology
Finding gene sets overlapping with a list of genes using Jaccard Similarity#
This query computes the Jaccard Similarity of a list of HUGO gene symbols, held one per line in a text file named members.txt, across all of the gene sets in MSigDB (here is an example file). Use MGI symbols if working with the Mouse database:
.import --schema test members.txt member_list
.mode tabs
.headers on
WITH QuerySet(member) AS (SELECT symbol FROM member_list)
SELECT standard_name, sum(InQuerySet) AS UnionCount,
(sum(NotInQuerySet) + (SELECT count(member) FROM QuerySet)) AS IntersectionCount,
CAST(sum(InQuerySet) AS REAL)/(sum(NotInQuerySet) +
(SELECT count(member) FROM QuerySet)) AS JaccSim
FROM ( SELECT standard_name,
CASE WHEN symbol IN ( SELECT member FROM QuerySet )
THEN 1 ELSE 0 END AS InQuerySet,
CASE WHEN symbol NOT IN ( SELECT member FROM QuerySet )
THEN 1 ELSE 0 END AS NotInQuerySet
FROM gene_set gset
INNER JOIN gene_set_gene_symbol gsgs ON gset.id = gsgs.gene_set_id
INNER JOIN gene_symbol gsym ON gsgs.gene_symbol_id = gsym.id )
GROUP BY standard_name ORDER BY JaccSim DESC LIMIT 20;
standard_name UnionCount IntersectionCount JaccSim
SOGA_COLORECTAL_CANCER_MYC_UP 79 170 0.464705882352941
WP_PYRIMIDINE_METABOLISM 24 227 0.105726872246696
KEGG_PURINE_METABOLISM 31 295 0.105084745762712
KEGG_PYRIMIDINE_METABOLISM 24 241 0.0995850622406639
GOBP_NUCLEOSIDE_MONOPHOSPHATE_BIOSYNTHETIC_PROCESS 18 191 0.0942408376963351
GOBP_RIBONUCLEOSIDE_MONOPHOSPHATE_BIOSYNTHETIC_PROCESS 16 185 0.0864864864864865
GOBP_NUCLEOSIDE_MONOPHOSPHATE_METABOLIC_PROCESS 19 225 0.0844444444444444
REACTOME_METABOLISM_OF_NUCLEOTIDES 20 244 0.0819672131147541
GOBP_RIBONUCLEOSIDE_MONOPHOSPHATE_METABOLIC_PROCESS 16 211 0.0758293838862559
REACTOME_NUCLEOTIDE_BIOSYNTHESIS 11 170 0.0647058823529412
GOBP_PURINE_NUCLEOSIDE_MONOPHOSPHATE_BIOSYNTHETIC_PROCESS 11 178 0.0617977528089888
MODULE_219 11 183 0.0601092896174863
SCHUHMACHER_MYC_TARGETS_UP 14 233 0.0600858369098712
GOBP_PURINE_NUCLEOSIDE_MONOPHOSPHATE_METABOLIC_PROCESS 11 201 0.054726368159204
GSE33292_WT_VS_TCF1_KO_DN3_THYMOCYTE_DN 19 348 0.0545977011494253
GOBP_NUCLEOSIDE_PHOSPHATE_BIOSYNTHETIC_PROCESS 24 440 0.0545454545454545
GOBP_GMP_BIOSYNTHETIC_PROCESS 9 172 0.0523255813953488
GOBP_RIBOSE_PHOSPHATE_BIOSYNTHETIC_PROCESS 20 385 0.051948051948052
MODULE_102 9 177 0.0508474576271186
GOBP_NUCLEOBASE_BIOSYNTHETIC_PROCESS 9 177 0.0508474576271186
Building the Legacy MSigDB XML File#
The following query will create the legacy deprecated MSigDB XML file, for any users that still need it. It will leave out a small amount of information related to the Hallmark collection gene sets, specifically the FOUNDER_NAMES, REFINEMENT_DATASETS, and VALIDATION_DATASETS. This info remains available from the GSEA website.
.output "msigdb_user.v2023.1.Hs.xml"
.headers off
.print '<?xml version="1.0" encoding="UTF-8"?>'
.print '<!-- Copyright (c) 2004-2023 Broad Institute, Inc., Massachusetts Institute of Technology, and Regents of the University of California. All rights reserved. -->'
SELECT format('<MSIGDB NAME="msigdb_user" VERSION="%s" BUILD_DATE="%s">',
version_name, version_date)
FROM MSigDB
WHERE version_name = '2023.1.Hs';
SELECT format(
' <GENESET STANDARD_NAME="%s" SYSTEMATIC_NAME="%s" HISTORICAL_NAME="" ORGANISM="%s" PMID="%s" AUTHORS="%s" GEOID="%s" EXACT_SOURCE="%s" GENESET_LISTING_URL="" EXTERNAL_DETAILS_URL="%s" CHIP="%s" CATEGORY_CODE="%s" SUB_CATEGORY_CODE="%s" CONTRIBUTOR="%s" CONTRIBUTOR_ORG="%s" DESCRIPTION_BRIEF="%s" DESCRIPTION_FULL="%s" TAGS="" MEMBERS="%s" MEMBERS_SYMBOLIZED="%s" MEMBERS_EZID="%s" MEMBERS_MAPPING="%s" FILTERED_BY_SIMILARITY="%s" FOUNDER_NAMES="" REFINEMENT_DATASETS="" VALIDATION_DATASETS=""/>',
standard_name, systematic_name, species_name, PMID, authorList, GEO_id,
exact_source, external_details_URL, ns.label,
iif(instr(collection_name, ':'), substr(collection_name,0,instr(collection_name,':')), collection_name),
iif(instr(collection_name, ':'), substr(collection_name,instr(collection_name,':')+1), ''),
contributor, contrib_organization,
description_brief, description_full,
memberList, symbolList, ezidList, mappingList, fbsTerms
)
FROM gene_set gset
INNER JOIN gene_set_details gsd ON gset.id = gsd.gene_set_id
INNER JOIN namespace ns ON primary_namespace_id = ns.id
INNER JOIN species sp ON ns.species_code = sp.species_code
INNER JOIN (
SELECT gene_set_id, group_concat(source_id) as memberList,
group_concat(symbol) as symbolList,
group_concat(NCBI_id) as ezidList,
group_concat(mapping,'|') as mappingList
FROM (
SELECT gssm.gene_set_id, source_id, symbol, NCBI_id,
source_id||','||symbol||','||NCBI_id AS mapping
FROM gene_set_source_member gssm
INNER JOIN source_member sm ON gssm.source_member_id = sm.id
INNER JOIN gene_symbol gsym ON sm.gene_symbol_id = gsym.id
ORDER BY gssm.gene_set_id, symbol) AS sym_info
GROUP BY gene_set_id ) AS syms ON syms.gene_set_id = gset.id
LEFT JOIN (
SELECT gene_set_id, group_concat(term) AS fbsTerms
FROM external_term_filtered_by_similarity
GROUP BY gene_set_id) AS extTerms ON extTerms.gene_set_id = gset.id
LEFT JOIN (
SELECT publication_id, PMID, group_concat(pub_auth.display_name, ',') AS authorList
FROM publication pub
INNER JOIN (
SELECT publication_id, display_name
FROM publication_author pa
INNER JOIN author au ON au.id = pa.author_id
ORDER BY publication_id, author_order
) AS pub_auth ON pub_auth.publication_id = pub.id
GROUP BY publication_id) AS pub_info ON pub_info.publication_id = gsd.publication_id
ORDER BY collection_name, lower(standard_name);
.print '</MSIGDB>'
.output
.headers on
About SQLite#
The official SQLite documentation is available at https://www.sqlite.org and an (unofficial) introductory tutorial is available at https://www.sqlitetutorial.net.
As a single-file database format, SQLite is well suited to our needs.
- It's self-contained https://www.sqlite.org/about.html
- It's not a networked client-server DB like MySQL, PostgreSQL, etc. so there is no additional set-up, administration, or maintenance in running the database.
- A database is held in a single file, matching the idea of a portable database analogous to our existing XML format.
- The “engine” is a small program (~1.1 MB) which reads local files.
- aside from initial installation, it’s ready to use directly.
- It has a full-featured SQL implementation.
- A relational model gives a better representation of MSigDB contents than XML can.
- It's very fast, especially compared to processing XML. The developers say it's "faster than the filesystem".
- It’s free and Open Source (Public Domain)
- It’s ubiquitous and widely used.
- There are programming language bindings for Python, R, Java (JDBC), Julia, C, etc.